Digital AssetAvailable
TBSP: Trajectory Inference Based on SNP information
Faculty of Medicine and Health Sciences
Biomedical Engineering
McGill University
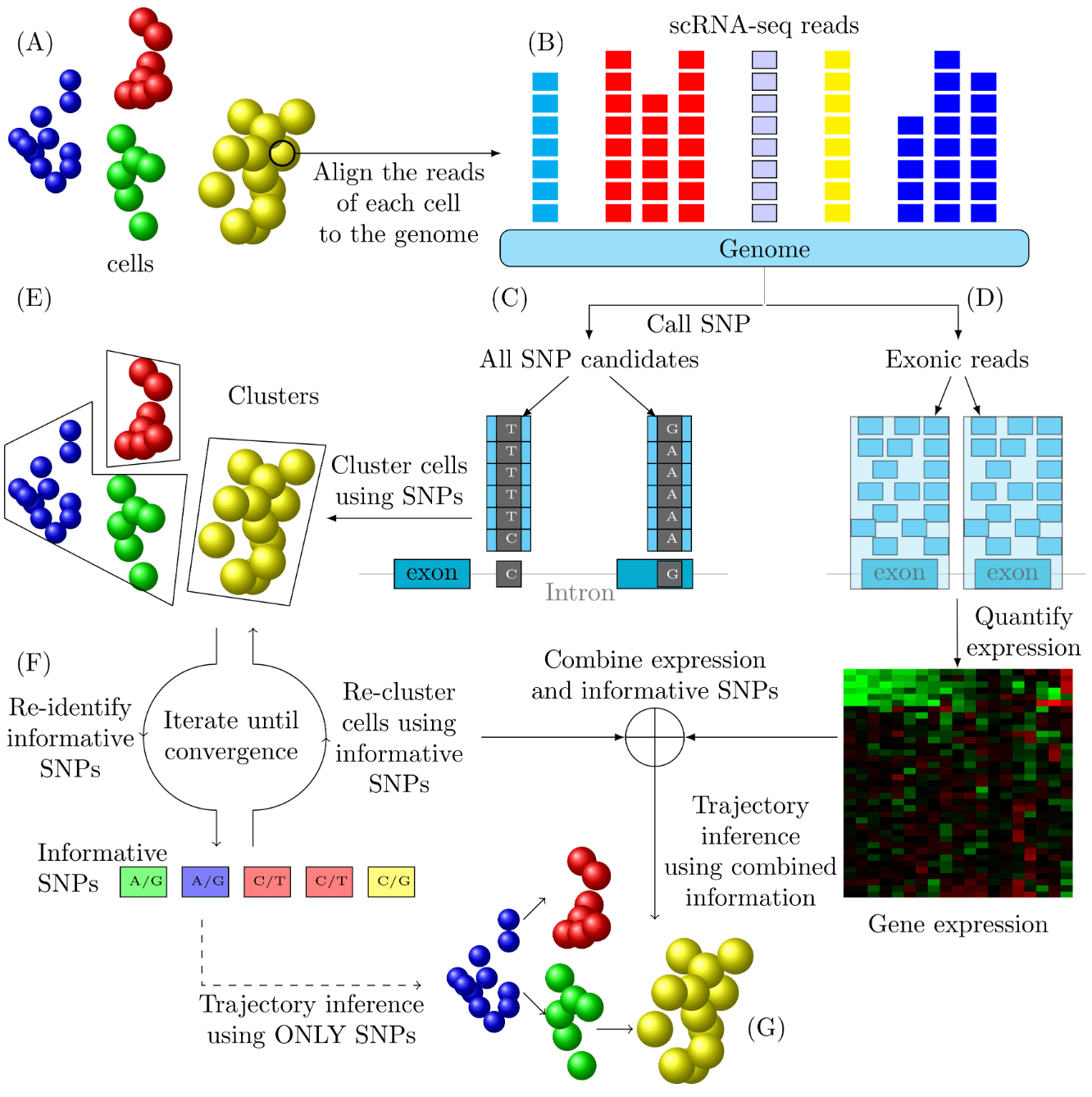
Several recent studies focus on the inference of developmental and response trajectories from single cell RNA-Seq (scRNA-Seq) data. A number of computational methods, often referred to as pseudo-time ordering, have been developed for this task. Recently, CRISPR has also been used to reconstruct lineage trees by inserting random mutations. However, both approaches suffer from drawbacks that limit their use. Here we develop a method to detect significant, cell type specific, sequence mutations from scRNA-Seq data. We show that only a few mutations are enough for reconstructing good branching models. Integrating these mutations with expression data further improves the accuracy of the reconstructed models.
Ding Lab
Faculty of Medicine and Health Sciences
Research lab focused on advancing scientific knowledge and innovation.
JD
Jun Ding
Digital AssetAvailable
TBSP: Trajectory Inference Based on SNP information
Faculty of Medicine and Health Sciences
Biomedical Engineering
McGill University
Several recent studies focus on the inference of developmental and response trajectories from single cell RNA-Seq (scRNA-Seq) data. A number of computational methods, often referred to as pseudo-time ordering, have been developed for this task. Recently, CRISPR has also been used to reconstruct lineage trees by inserting random mutations. However, both approaches suffer from drawbacks that limit their use. Here we develop a method to detect significant, cell type specific, sequence mutations from scRNA-Seq data. We show that only a few mutations are enough for reconstructing good branching models. Integrating these mutations with expression data further improves the accuracy of the reconstructed models.
Ding Lab
Faculty of Medicine and Health Sciences
Research lab focused on advancing scientific knowledge and innovation.
JD
Jun Ding
You might also like
Discover more resources that could support your research